The quick answer to this question is a Common Specification (CS):
“means a set of technical and/or clinical requirements, other than a standard, that provides a means of complying with the legal obligations applicable to a device, process or system.”1
Essentially, the European Commission has reserved the right within both EU MDR1 and EU IVDR5 to develop Common Specifications for certain groups of devices where harmonized standards do not exist or where they are insufficient to provide a means of complying with specific General Safety and Performance Requirements (GSPRs).
In addition to technical requirements, Common Specifications may also cover requirements such as the application of risk management, clinical investigations and clinical evaluation, and/or post-market clinical follow-up.
As of February 23, 2023, the European Commission has published three implementing regulations that lay down common specifications for:
- certain Class D in vitro diagnostic medical devices2;
- groups of products without an intended medical purpose3; and
- the reprocessing of single-use devices4.
So, if your organization markets medical devices within these categories, you must determine whether there are Common Specifications that apply to your device.
If so, you should demonstrate conformance with applicable Common Specifications to comply with specific General Safety and Performance Requirements (GSPRs). Showing conformity with GSPRs is a prerequisite to a medical device being CE Marked.
It is important to note that for devices without an intended medical purpose, in the preamble to EU MDR1, Section 12, the European Commission clarifies that:
“Such common specifications should be developed specifically for a group of products without an intended medical purpose and should not be used for conformity assessment of the analogous devices with a medical purpose. Devices with both a medical and a non-medical intended purpose should fulfil both the requirements applicable to devices with, and to devices without, an intended medical purpose.”
With this, the European Commission makes it clear that devices with a medical purpose should be using harmonized standards and/or a non-harmonized standard approach to show conformance to GSPRs.
If you are looking for information regarding the role of harmonized standards under EU MDR and EU IVDR and their importance as part of the CE conformity assessment process for medical devices or in vitro diagnostic medical devices please see this link to my blog post on this subject titled What Are Harmonized Standards Under EU MDR And EU IVDR?
In the next three sections, I’ll provide a few more details regarding the scope of the Common Specifications discussed earlier that have been published to date.
Certain Class D in vitro diagnostic medical devices (Regulation (EU) 2022/1107) 2
This regulation lays out common specifications for in vitro diagnostic medical devices intended for the detection and/or quantification of markers for the following:
- Blood group antigens in the ABO, Rh, Kell, Duffy and Kidd blood group systems
- Human immunodeficiency virus (HIV) infection
- human T-cell lymphotropic virus (HTLV) infection
- Hepatitis C virus (HCV) infection
- Hepatitis B virus (HBV) infection
- Hepatitis D virus (HDV) infection
- Variant Creutzfeldt-Jakob disease (vCJD)
- Cytomegalovirus (CMV) infection
- Epstein-Barr virus (EBV) infection
- Treponema pallidum (T. pallidum)
- Trypanosoma cruzi (T. cruzi) infection
- Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection
Groups of products without an intended medical purpose (Regulation (EU) 2022/2346)3
This regulation lays out common specifications for groups of products without an intended medical purpose listed in Annex XVI of Regulation (EU) 2017/745 (EU MDR) and specifically includes:
- Contact lenses
- Products intended to be totally or partially introduced into the human body through surgically invasive means for the purpose of modifying the anatomy, with the exception of tattooing products and piercings
- Substances, combinations of substances, or items intended to be used for facial or other dermal or mucous membrane filling by subcutaneous, submucous or intradermal injection or other introduction, excluding those for tattooing
- Equipment intended to be used to reduce, remove or destroy adipose tissue, such as equipment for liposuction, lipolysis or lipoplasty
- High intensity electromagnetic radiation (for example, infra-red, visible light and ultra-violet) emitting equipment intended for use on the human body, including coherent and non-coherent sources, monochromatic and broad spectrum, such as lasers and intense pulsed light equipment, for skin resurfacing, tattoo or hair removal or other skin treatment
- Equipment intended for brain stimulation that apply electrical currents or magnetic or electromagnetic fields that penetrate the cranium to modify neuronal activity in the brain
Reprocessing of single-use devices (Regulation (EU) 2020/1207)4
This regulation gives requirements for reprocessing of single-use devices where permitted by national law.
Is compliance with Common Specifications a requirement?
Based on 2017/745, Chapter I, Article 9, as quoted below manufacturers do not need to comply with applicable Common Specifications but should document justification regarding why the solution, they have chosen to employ is acceptable. They should also be prepared for additional questions during the conformity assessment process by notified bodies or competent authorities.
“Manufacturers shall comply with the CS referred to in paragraph 1 unless they can duly justify that they have adopted solutions that ensure a level of safety and performance that is at least equivalent thereto.”
Where to find Common Specifications for MDR and IVDR?
Like other EU Regulations, Common Specifications may be updated, repealed, or new ones released over time. This is why it is very important to periodically go to the source which is the EUR-Lex website which is an official website of the European Union and provides access to European law.
EUR-LEX Web address: https://eur-lex.europa.eu/homepage.html
How to search EUR-Lex Website for Common Specifications?
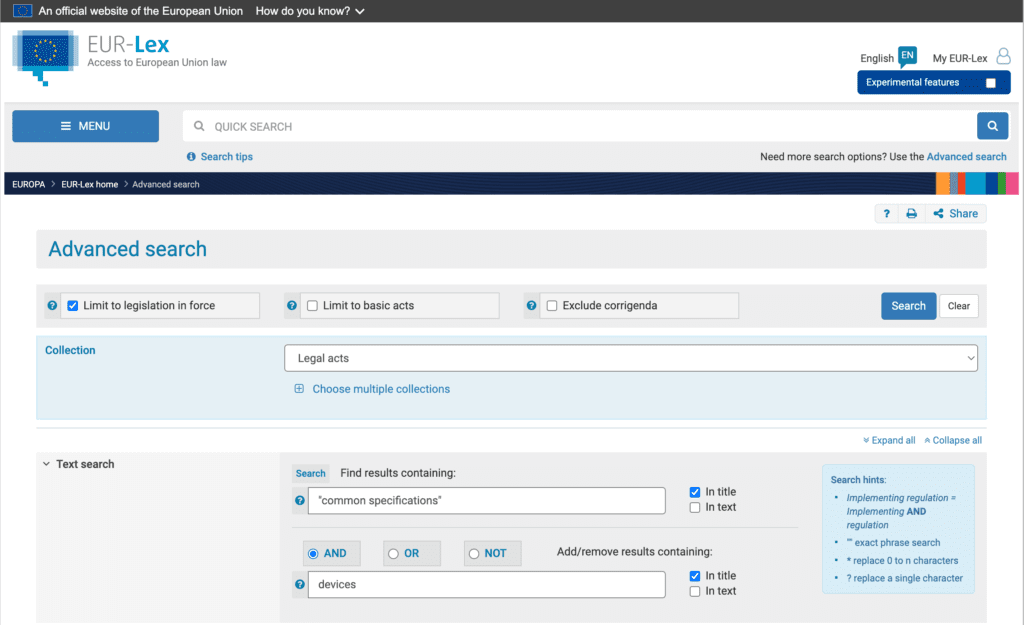
There are many documents within the EUR-Lex database and using the Quick Search option can result in many results that are difficult to navigate. Therefore, I recommend using the Advanced Search option (the link is just below the search button).
I have had success setting the following Advanced Search parameters:
Check Box: Limit to legislation in force
Collection: Legal acts
Text search: Find results containing “common specifications” and devices
This limits the results just to Common Specifications related to medical devices and regulation that is currently in force. Go ahead and play around with the options to expand the search to documents that may be under review or not in force if this is of interest to you.
Summary
Common Specifications are technical and/or clinical requirements, other than a standard, that provides a means of complying with the legal obligations applicable to a device, process or system. They provide a pathway of complying with specific General Safety and Performance Requirements (GSPRs) for certain groups of devices where harmonized standards do not exist or where they are insufficient to meet EU Law.
Common Specifications, in addition to technical requirements, may also cover requirements related to the application of risk management, clinical investigations and clinical evaluation, and/or post-market clinical follow-up.
Although complying with Common Specifications, if applicable, is not mandatory they are usually the first choice for demonstrating conformity to GSPRs as part of the CE Marking conformity assessment process. Although alternative methods are possible, they come with the additional task of justifying the approach sufficiently demonstrates conformance to GSPRs.
References:
1REGULATION (EU) 2017/745 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 5 April 2017 on medical devices, amending Directive 2001/83/EC, Regulation (EC) No 178/2002 and Regulation (EC) No 1223/2009 and repealing Council Directives 90/385/EEC and 93/42/EEC
2COMMISSION IMPLEMENTING REGULATION (EU) 2022/1107 of 4 July 2022 laying down common specifications for certain class D in vitro diagnostic medical devices in accordance with Regulation (EU) 2017/746 of the European Parliament and of the Council
3COMMISSION IMPLEMENTING REGULATION (EU) 2022/2346 of 1 December 2022 laying down common specifications for the groups of products without an intended medical purpose listed in Annex XVI to Regulation (EU) 2017/745 of the European Parliament and of the Council on medical devices
4COMMISSION IMPLEMENTING REGULATION (EU) 2020/1207 of 19 August 2020 laying down rules for the application of Regulation (EU) 2017/745 of the European Parliament and of the Council as regards common specifications for the reprocessing of single-use devices
5REGULATION (EU) 2017/746 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 5 April 2017 on in vitro diagnostic medical devices and repealing Directive 98/79/EC and Commission Decision 2010/227/EU